

Breaking through treatment bottlenecks! A new journey begins in the development of innovative therapy for Gaucher disease
Gaucher Disease (GD) is a rare autosomal recessive lysosomal storage disorder caused by defects in the GBA gene, leading to a deficiency of glucocerebrosidase activity, which results in the abnormal accumulation of glucocerebroside within macrophages, affecting multiple organs such as the liver, spleen, bone marrow, and central nervous system, severely threatening the life and health of patients. According to the involvement of the nervous system, Gaucher Disease is mainly divided into three types: Type 1 GD (non-neuronopathic) is the most common and only affects the viscera and bone marrow; Type 2 GD (acute neuronopathic) has a severe onset, beginning in infancy with rapid progression, and most patients die within 2 years; Type 3 GD (neuronopathic visceral) begins in childhood or adolescence, involving both visceral damage and progressive neurological disorders, with a moderate prognosis that worsens as neurological symptoms progress. Statistics show that the global incidence of Gaucher Disease is only 1 in 100,000, and the prevalence in the Chinese population is only 1 in 500,000 to 1 in 200,000. It has been included in China's "First Batch of Rare Disease Catalog".
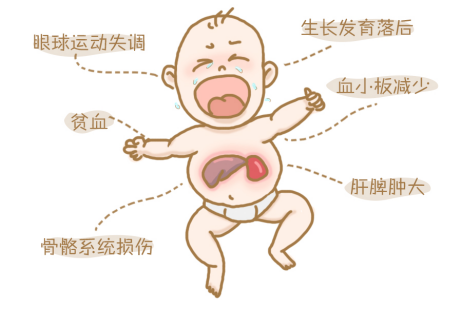
图源于网络
For a long time, the mainstream clinical treatments for Gaucher disease have been enzyme replacement therapy (ERT) and substrate reduction therapy (SRT), but there are many limitations: ERT requires long-term intravenous infusion, has low compliance, and cannot improve GD3-type neurological lesions; SRT has limited efficacy, is difficult to control long-term disease progression, and some patients cannot tolerate it. In recent years, with the iteration of pharmaceutical research and development technologies, there has been an explosion in innovative drug development for Gaucher disease. Oral targeted drugs, gene therapies, and other medications have emerged, covering different subtypes, allowing treatment to steadily progress from 'symptomatic relief' to 'precise cure,' bringing new hope to patients.
Core Breakthrough: Direct Hit on GD3-Type Neuropathy
Progressive neurological lesions in type GD3 are the biggest clinical bottleneck in the treatment of Gaucher disease. Traditional ERT drugs cannot cross the blood-brain barrier, making it difficult to alleviate neurological symptoms such as ataxia and cognitive decline, which can ultimately lead to the patient losing the ability to live independently. In February 2026, Sanofi announced the results of the phase III LEAP2MONO clinical trial of Venglustat. This innovative drug has become the first to show superiority over traditional ERT therapy in improving neurological symptoms, and it is expected to fill the global gap in the treatment of neurological lesions in type GD3.
Venglustat is a novel oral glucosylceramide synthase inhibitor (GCSi), whose core advantage lies in its good blood-brain barrier penetration, allowing it to directly enter the central nervous system to fundamentally inhibit the abnormal synthesis and accumulation of glucosylceramide, while also addressing systemic symptom control. Additionally, the once-daily oral administration greatly improves patient adherence, freeing them from the hassle of long-term intravenous infusion associated with traditional ERT drugs.

This phase III clinical trial was a double-blind, double-dummy, active-controlled study that enrolled 43 GD3 patients aged 12 years and older who had received at least 3 years of ERT treatment and had achieved systemic symptom targets. Participants were randomized 1:1 to the oral Venglustat group or the intravenous ERT control group. After 52 weeks of treatment, the Venglustat group achieved all predefined primary endpoints, with neurological function assessment showing significantly better efficacy compared to ERT therapy, while the control of systemic organs and hematologic indicators was comparable to standard treatment.
Based on this positive result, Sanofi plans to submit a global marketing authorization application for Venglustat for type 3 Gaucher disease. The drug has received orphan drug designation in Europe, the U.S., and Japan, and has also obtained fast track designation from the U.S. FDA. The success of its Phase III clinical trial not only brings new hope to patients with type 3 Gaucher disease, but also provides an important reference for the development of other lysosomal storage disease drugs associated with neurological involvement.
AAV gene breakthrough in the treatment limitations of GD1 type
In addition to breakthroughs in oral targeted drugs, gene therapy as a 'curative' treatment has also made significant progress. Recently, at the 22nd WORLD Symposium held in San Diego, USA, Spur Therapeutics announced the latest data from its Phase I/II clinical study of the investigational AAV gene therapy FLT201 for the treatment of type I Gaucher disease.
FLT201 uses a self-developed liver-targeted AAVS3 vector, carrying an engineered, more stable β-glucocerebrosidase variant functional gene. It can continuously supplement the missing glucocerebrosidase in patients' stem cells, efficiently degrade abnormally accumulated glycolipids and toxic metabolites, address the enzyme deficiency problem at its root, and is expected to achieve the therapeutic goal of 'one-time treatment, long-term benefit.'

FLT201载体设计
The data announced this time come from the GALILEO-1 Phase I/II trials and the GALILEO-2 long-term follow-up study, with enrolled patients all having received at least 2 years of standard ERT/SRT treatment. Among them, 4 patients who received a single low-dose treatment of FLT201 discontinued their original therapy within 11 weeks, and as of the data cutoff in November 2025, have remained off treatment for 20-26 months. After treatment, patients' key biomarkers and clinical indicators showed sustained improvement: lysosomal glucosylceramide significantly decreased by 53%-94% during the two-year follow-up; the challenging issue of bone marrow substrate accumulation was notably addressed, with some patients achieving complete clearance of this indicator; hemoglobin, platelet counts, and liver and spleen volumes all remained improved or stable, allowing patients to completely avoid the frequent treatment burden of traditional therapy.
Based on excellent Phase I/II data, Spur has initiated a global multicenter Phase III clinical trial of FLT201, and has reached a consensus with the U.S. FDA on the single-arm trial design. This achievement is expected to support both accelerated and full approval of the drug, laying a solid foundation for its early market launch.
Domestic ERT new drug approved for market, filling domestic gap
While oral targeted drugs and gene therapy steadily advance, domestic enzyme replacement therapy (ERT) new drugs have also achieved major breakthroughs. Vimizim® (velaglucerase beta for injection), developed by Beihai Kangcheng, was approved for marketing by the NMPA on May 15, 2025, becoming China's first new ERT drug for Gaucher disease. It is suitable for the long-term treatment of adolescents aged 12 and above and adults with type 1 and type 3 Gaucher disease, filling a gap in the domestic field, breaking the monopoly of imported ERT drugs, and greatly improving treatment accessibility.
The launch of Gorening® is highly milestone-significant, with its research and approval process being efficiently advanced throughout: In August 2024, the positive topline data of the pivotal clinical trial was announced; in September, it obtained priority review qualification from the CDE; in November, the new drug marketing application was formally accepted; in March 2025, it passed registration verification and pre-marketing GMP compliance inspections; and in May, it was officially approved, highlighting the accelerated progress of rare disease drug development and approval in China.
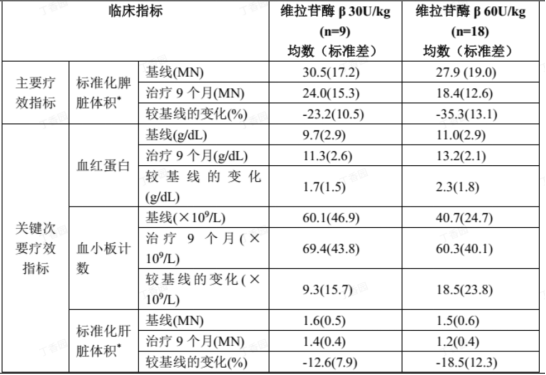
The pivotal clinical trial of this drug was a randomized, double-blind, dose-comparison study that evaluated the efficacy, safety, and pharmacokinetics of intravenous infusion every two weeks in treatment-naive patients, and included an open-label extension period. The results showed that both the 60 U/kg and 30 U/kg dose groups successfully achieved the primary efficacy endpoints. After 9 months of treatment, subjects' spleen volume was reduced on average from baseline, with efficacy not inferior to imported ERT drugs, and good safety. As a first-in-class innovative drug, Gorenin® has accumulated comprehensive data throughout the entire development chain. After market launch, it will significantly reduce treatment costs, break the price barriers of imported drugs, allow more ordinary families to afford standard treatment, and truly achieve universal access to Gaucher disease therapy.
In the future, the treatment of Gaucher disease will continue to develop toward precision, long-lasting effects, and universal accessibility. Breakthroughs in targeted and gene therapies will further overcome the limitations of traditional treatments, promoting a shift from symptomatic relief to root-cause therapies. At the same time, the introduction and iteration of domestically developed innovative drugs will continuously improve drug accessibility, reduce treatment costs, and gradually establish a high-quality treatment system covering different subtypes. This will also provide valuable experience for the research and development of diagnosis and treatment for other rare diseases, bringing better treatment prospects to more patients with rare diseases.