Macrophage ADAM8: A Key Inhibitory Target for Post-Myocardial Infarction Repair
"Sudden chest tightness as if crushed by a boulder, struggling to breathe..." This is a common intuitive experience of myocardial infarction (MI). As a globally recognized "cardiovascular killer," MI not only has a high mortality rate but also leaves many survivors with sequelae such as heart failure and arrhythmia, significantly impairing their quality of life. As a major disease burden in the global cardiovascular field, MI's high mortality and morbidity have long been core challenges in clinical diagnosis and treatment. Recently, a study by Chinese scientists published in the international journal Journal of Advanced Research has brought new hope for MI treatment: they identified ADAM8 in macrophages as the "key culprit" hindering cardiac repair post-MI. This discovery not only clarifies the critical barrier to cardiac repair but also provides a clear direction for the development of novel drugs.
I. Myocardial Infarction: More Than Just "Heart Pain"
According to the World Health Organization (WHO), approximately 17.9 million people worldwide die of cardiovascular diseases each year, a significant proportion of which are caused by myocardial infarction (MI). In China, the incidence of MI has been on the rise annually, with one death from cardiovascular diseases occurring every 18-20 seconds, presenting an extremely grim situation.
What exactly is myocardial infarction? Briefly, MI refers to myocardial necrosis caused by acute and persistent myocardial ischemia and hypoxia due to coronary artery occlusion. The coronary arteries are critical blood vessels responsible for supplying blood to the heart. Once occluded, the cardiac muscle begins to die from insufficient oxygen and nutrients—analogous to "cutting off the food supply" to the heart, with catastrophic consequences.
Most people's understanding of MI is confined to "chest pain", but its clinical manifestations are far more complex than perceived. The typical symptom is oppressive or constrictive pain in the retrosternal region or precordium, persisting for several minutes without relief. Atypical symptoms are more insidious: for instance, some elderly patients and those with diabetes may only present with chest tightness, dyspnea, or even toothache, jaw pain, and upper abdominal pain, which are highly prone to misdiagnosis. Additionally, some individuals may experience "prodromal symptoms" such as frequent chest tightness in the days preceding the acute attack, which exacerbates with physical activity. Prompt medical consultation is imperative in such cases to prevent adverse outcomes.
Given the severe morbidity and mortality associated with MI, current clinical treatment strategies are centered on the core objectives of "revascularization, myocardial protection, and secondary prevention", forming a comprehensive therapeutic system integrating pharmacotherapy with interventional/surgical interventions:
Drug Category | Specific Drugs | Core Mechanisms |
Thrombolytic Agents | Urokinase, Streptokinase, Recombinant Human Tissue-Type Plasminogen Activator (Alteplase) | Activate the fibrinolytic system to dissolve intravascular thrombi and achieve revascularization. |
Antiplatelet Agents | Aspirin, Clopidogrel, Ticagrelor | Inhibit platelet aggregation to prevent thrombosis, particularly indicated for post-stent implantation thrombosis prophylaxis. |
Anticoagulants | Heparin, Low-Molecular-Weight Heparin | Block the coagulation cascade to assist in thrombosis prevention; typically used short-term during hospitalization. |
Statins | Atorvastatin, Rosuvastatin | Regulate lipid metabolism, stabilize atherosclerotic plaques, and reduce the risk of MI recurrence. |
β-Blockers | Metoprolol | Improve myocardial remodeling and decrease the incidence of post-infarction heart failure. |
Angiotensin-Converting Enzyme Inhibitors (ACEIs) | Benazepril, etc. | Improve myocardial remodeling and reduce the risk of post-infarction heart failure. |
Angiotensin Ⅱ Receptor Blockers (ARBs) | Valsartan, etc. | Share a similar mechanism to ACEIs; indicated for patients intolerant to ACEIs to improve myocardial remodeling. |
Despite the significant improvement in acute-phase revascularization rates achieved by interventional therapy and coronary artery bypass grafting (CABG), current therapeutic strategies still fail to effectively address the core challenge of post-infarction myocardial repair—specifically, how to promote angiogenesis in necrotic myocardial regions and inhibit excessive fibrosis. This unmet clinical need has emerged as a key breakthrough point in cardiovascular drug development in recent years, highlighting an urgent demand for identifying critical molecular targets that regulate the myocardial repair process. Consequently, researchers have been actively pursuing novel targets capable of fundamentally promoting cardiac repair, and a recent study published in Journal of Advanced Research has identified such a pivotal molecule.
II. New Hope: Identifying the "Key Molecule" Hindering Cardiac Repair
In the process of cardiac repair, macrophages act as both "cleaners" and "repairers" in the body. Following myocardial infarction, they rapidly migrate to the necrotic area to clear dead cardiomyocytes and debris, while secreting various factors to promote angiogenesis and tissue repair. The core finding of this study is that ADAM8—a "saboteur" within macrophages—transforms these repair-oriented "workers" into "destroyers."
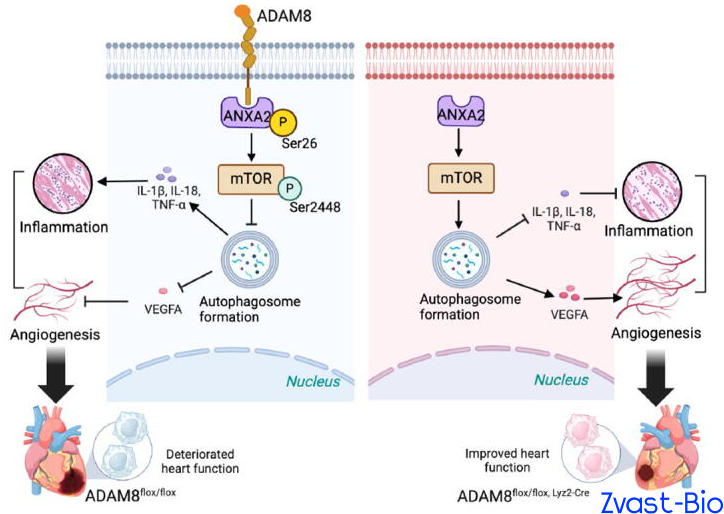
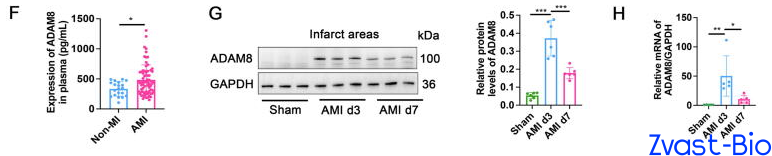
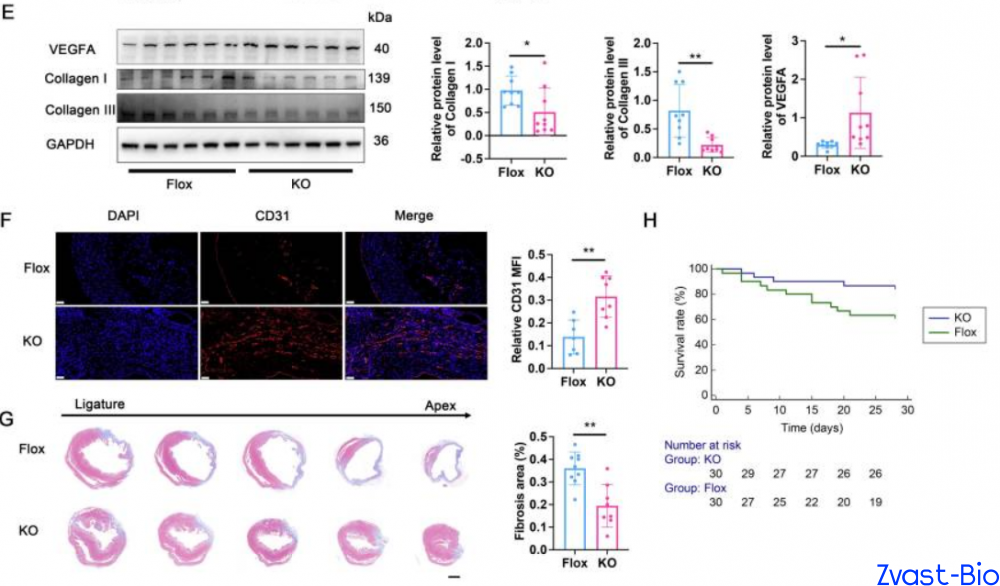
The research team first validated through clinical data that plasma ADAM8 levels in patients with acute myocardial infarction (AMI) were significantly higher than those in non-MI populations. Subsequent experiments in mice revealed that ADAM8 expression in cardiac macrophages peaked on day 3 post-MI, with predominant localization within macrophages and minimal expression in other cell types—indicating a potential close association between ADAM8 and the pathological progression of post-MI. To verify the "destructive role" of ADAM8, researchers generated macrophage-specific ADAM8 knockout mice. Results demonstrated that following MI, these knockout mice exhibited significantly enhanced cardiac repair capacity, with a substantially higher 28-day survival rate compared to wild-type mice. Conversely, macrophage-specific overexpression of ADAM8 in mice led to a drastic deterioration in post-MI repair outcomes. These findings further confirm that ADAM8 is indeed a key molecule hindering cardiac repair.
III. Mechanistic Insights: How ADAM8 Exerts Its "Destructive Effects"
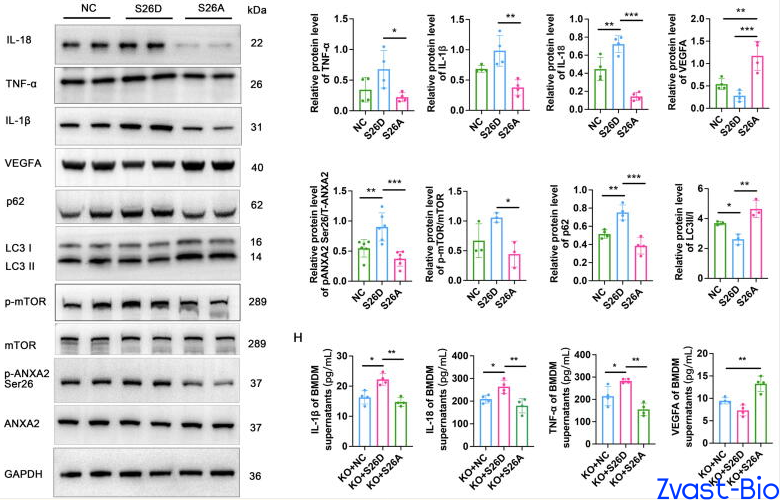
In-depth investigations uncovered the underlying mechanism by which ADAM8 mediates cardiac repair impairment: it binds to the ANXA2 protein, promoting phosphorylation at its Ser26 site, which in turn activates the mTOR signaling pathway and ultimately inhibits autophagy. Autophagy, a critical process for cellular debris clearance and homeostasis maintenance, is indispensable for normal macrophage function.
ADAM8-induced autophagy inhibition triggers a phenotypic switch in macrophages: increased secretion of pro-inflammatory cytokines (e.g., IL-1β, TNF-α) exacerbates local inflammation in the infarcted area, while reduced production of pro-angiogenic factors (e.g., VEGFA) impairs neovascularization. This reciprocal dysregulation directly hinders cardiac repair and aggravates myocardial damage. Conversely, ADAM8 knockout blocks this "destructive pathway," reactivating autophagy and shifting macrophages toward an "anti-inflammatory and pro-reparative" phenotype that facilitates cardiac recovery.
IV. Clinical Translational Value of ADAM8 as a Therapeutic Target
This study provides two core directions for drug development in myocardial infarction (MI) treatment:
ADAM8 as a potential biomarker for MI diagnosis.Given the specific elevation of plasma ADAM8 levels in patients with acute MI, ADAM8-based diagnostic kits could be developed to support early clinical diagnosis. This approach is particularly applicable to suspected cases with atypical symptoms, thereby improving diagnostic accuracy.
ADAM8 Inhibitors hold defined drug development potential.The ADAM8-specific inhibitor BK-1361 used in this study has demonstrated significant myocardial protective effects in animal models. Future optimization of drug formulation and enhancement of targeting capabilities are expected to lead to the development of novel targeted therapeutics for post-MI repair. Unlike traditional revascularization agents, ADAM8 inhibitors improve the myocardial repair process at its root by regulating macrophage function, promoting angiogenesis, and inhibiting fibrosis—with the potential to reduce the incidence of post-infarction heart failure.
References:Ji Z, Guo J, Zhang R, Zuo W, Xu Y, Qu Y, Tao Z, Li X, Li Y, Yao Y, Ma G. ADAM8 deficiency in macrophages promotes cardiac repair after myocardial infarction via ANXA2-mTOR-autophagy pathway. J Adv Res. 2025 Jul;73:483-499. doi: 10.1016/j.jare.2024.07.037. Epub 2024 Aug 2. PMID: 39097092; PMCID: PMC12225930.
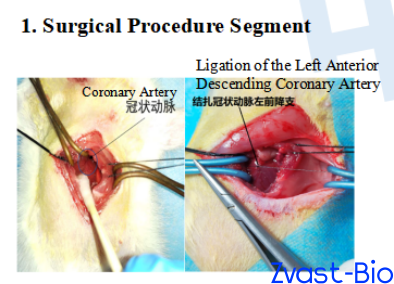
SD Rat Myocardial Infarction Model (Left Anterior Descending Coronary Artery Ligation)
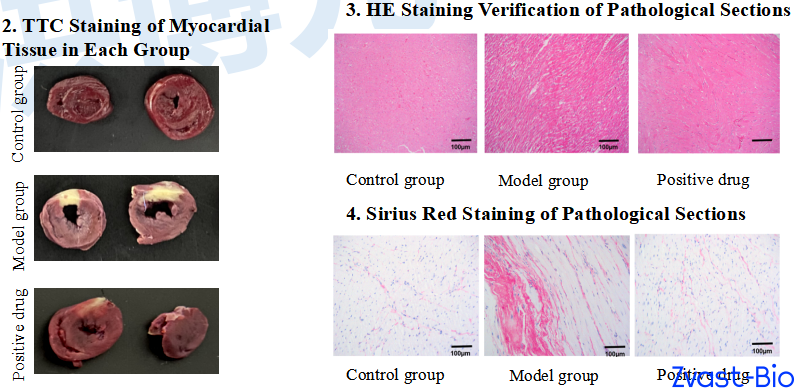
Modeling:After anesthetization, SD rats were fixed supine on the operating table. A midline cervical incision was made to dissect tissues and expose the trachea, which was then intubated and connected to a ventilator. An electrocardiograph was attached for continuous monitoring. The thoracic cavity was opened, and the pericardium was incised to expose the heart. A 6-0 silk suture with a round needle was used to ligate the left anterior descending coronary artery (LAD) at the upper-middle 1/3 segment. Successful modeling was confirmed by electrocardiographic changes, followed by thoracic cavity closure and intramuscular injection of penicillin for infection prophylaxis.Sham Operation Group: The same surgical procedure was performed, including threading at the identical LAD site, but without ligation.
Positive drug:Perindopril